

Sequenciamento do genoma coloca Embrapa na vanguarda do uso de ferramentas genômicas para melhoramento de forrageiras tropicais, recurso já usado em culturas como soja, milho e eucalipto.
Com o uso de tecnologias de sequenciamento de DNA e bioinformática de última geração, pesquisadores da Embrapa montaram o genoma da Urochloa ruziziensis, uma das gramíneas forrageiras mais utilizadas na agropecuária tropical, especialmente em sistemas de integração. As informações geradas a partir do genoma dessa espécie de braquiária darão suporte ao desenvolvimento de cultivares forrageiras de forma mais precisa e dinâmica. Dessa forma, os programas de melhoramento genético de forrageiras poderão solucionar problemas da agropecuária com maior eficiência e agilidade ao disponibilizarem cultivares melhoradas em menor tempo.
Marco Pessoa Filho, pesquisador da Embrapa Cerrados (DF), explica que a U. ruziziensis tem proximidade evolutiva com as demais espécies do gênero Urochloa mais utilizadas nas pastagens tropicais cultivadas – U. brizantha, U. decumbens e U. humidicola. Mas enquanto essas espécies são apomíticas, ou seja, se reproduzem de forma assexuada, gerando sementes clonais sem troca de material genético, a U. ruziziensis tem modo de reprodução sexual, por meio da troca de pólen.
Forrageira presente em mais de 80% das pastagens brasileiras
Nativos da África, os capins braquiária, atualmente classificados como do gênero Urochloa, são os mais cultivados no mundo tropical, sendo que no Brasil ocupam mais de 80% da área de pastagens cultivadas, segundo estimativas. Por outro lado, poucas cultivares são usadas nos plantios – daí a necessidade de disponibilização de novos genótipos para diversificar a genética das pastagens e diminuir a vulnerabilidade dos sistemas pastoris no País quanto a estresses bióticos, como pragas e doenças, e abióticos, como variações climáticas.
O problema é que os programas de melhoramento genético de espécies forrageiras necessitam de dez a 15 anos para desenvolver e lançar uma nova cultivar. A aplicação de métodos que usam informação genômica, como seleção genômica e seleção assistida por marcadores moleculares, tem o potencial de aumentar as taxas de ganho genético e reduzir os ciclos de seleção, tornando os programas de melhoramento mais rápidos.
O melhoramento genético apoiado por ferramentas genômicas já é realidade em culturas como soja, milho e eucalipto. Com o sequenciamento genômico da U. ruziziensis, a Embrapa pode ser pioneira na aplicação dessas ferramentas em melhoramento de forrageiras tropicais, inserindo essas espécies na era da genômica. 
Por essa característica, uma vantagem da U. ruziziensis, segundo o pesquisador, é a possibilidade de recombinar a diversidade genética da espécie por cruzamentos e de selecionar as melhores combinações por melhoramento genético clássico, sem as limitações das espécies apomíticas, cujas cultivares são geralmente obtidas a partir de acessos (amostras coletadas no campo e que representam a variação genética de uma população ou de um indivíduo propagado por clones), o que limita a variabilidade genética; ou por cruzamento entre diferentes espécies, o que em alguns casos pode resultar em problemas de compatibilidade e produção de sementes.
Além disso, a U. ruziziensis tem genoma diploide, ou seja, o DNA da planta está organizado em dois conjuntos de cromossomos (no caso dessa espécie, nove pares), enquanto as demais braquiárias cultivadas são poliploides, apresentando quatro, seis ou mais conjuntos de cromossomos. A espécie também foi escolhida para estudo por ter um genoma relativamente pequeno. “É um genoma mais simples de trabalhar quando comparado a outras braquiárias”, explica Pessoa.
“Ao melhorarmos geneticamente a U. ruziziensis, podemos tanto obter bons materiais para o desenvolvimento de cultivares dessa espécie (recombinação intraespecífica) como também apoiar o melhoramento das espécies apomíticas, fornecendo bons indivíduos sexuais que poderão ser cruzados com bons indivíduos apomíticos (recombinação interespecífica)”, acrescenta o pesquisador.
Ele lembra que a redução nos custos de novas tecnologias de genômica tem permitido aos grupos de pesquisa atuarem de forma independente, sem a necessidade do estabelecimento de grandes redes. No caso da montagem do genoma da U. ruziziensis, a equipe contou com a prestação de serviços de sequenciamento externos, realizando a maior parte do trabalho computacional na própria Embrapa.
Sequenciamento do genoma de Urochloa ruziziensis
Amostras foram enviadas ao Centro de Inovação Genome Quebec, na Universidade McGill, no Canadá, onde o material genético foi lido e “traduzido” para um extenso texto com sequências de letras que representam as bases nitrogenadas (adenina, guanina, citosina e timina) que compõem a molécula do DNA. O projeto utilizou tecnologia de sequenciamento de terceira geração (veja os passos no desenho abaixo), baseada em longos fragmentos de DNA, em equipamento PacBio Sequel.
Esse texto com dados brutos recebeu correções e foi montado com o uso do software Falcon-Unzip, gerando um primeiro rascunho do genoma, composto por milhares de sequências chamadas contigs. “São grandes fragmentos de DNA, mas ainda sem nenhuma informação sobre sua localização, por exemplo, em cromossomos”, diz Pessoa. 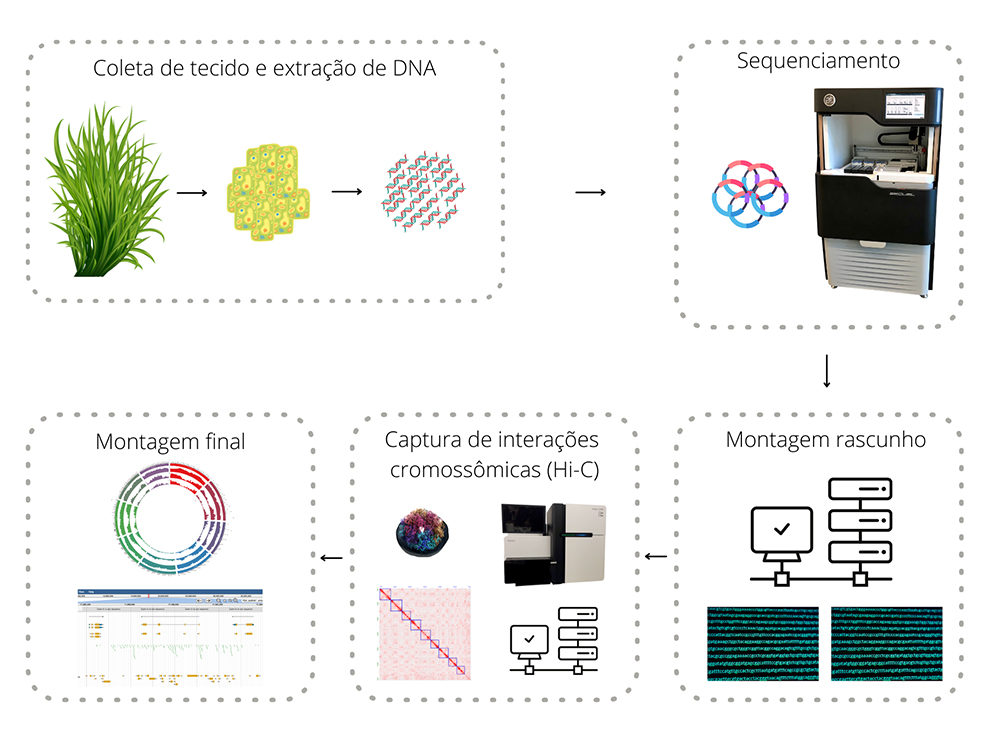
Com essas informações, foi possível montar a primeira versão do genoma da gramínea forrageira, em escala cromossômica, com tamanho de 604 milhões de pares de bases nitrogenadas (Mpb), representando 98% do tamanho total, estimado em cerca de 615 Mpb. O modelo estrutural obtido pela pesquisa contém 2.348 contigs organizados em nove scaffolds cromossômicos.
Em seguida, foi realizado um estudo de predição de genes na montagem, utilizando dados de sequenciamento de RNA (ácido ribonucleico, molécula formada a partir do DNA que atua na síntese de proteínas, expressando as informações do DNA) ou RNAseq a partir de cinco tecidos – folha, haste, haste reprodutiva, inflorescência e raiz – do mesmo clone de U. ruziziensis utilizado no sequenciamento do genoma. Dados de RNAseq de outros 11 acessos de U. ruziziensis disponíveis em bancos de dados públicos também foram utilizados nessa etapa.
O mapeamento desse conjunto de dados no genoma auxiliou na identificação de cerca de 40 mil modelos gênicos – regiões do genoma correspondentes aos genes e que representam, em tese, a porção “funcional” do genoma. Eles são identificados por softwares que procuram padrões típicos de regiões gênicas em sequências de DNA, além de usarem informações adicionais, como as geradas pelo RNAseq.